ЗАКОНОМЕРНОСТИ НАСЛЕДОВАНИЯ ПРИЗНАКОВ ЧЕЛОВЕКА И МЕТОДЫ ИХ ИЗУЧЕНИЯ
ГЕНЕАЛОГИЧЕСКИЙ МЕТОД
При изучении наследования нормальных и патологических признаков человека используют различные методы. Наиболее универсальным является генеалогический, т. е. метод родословных. Его сущность заключается в прослеживании передачи признака (болезни) среди родственников больного в нескольких поколениях. В медицинской генетике метод чаще называют клинико-генеалогическим, поскольку речь идет об изучении патологических признаков в семье с помощью приемов клинического обследования. В отличие от морфологических, иммунологических, биохимических и прочих специальных методов, требующих подчас сложного оборудования и длительного кропотливого лабораторного анализа, клинико-генеалогический метод относительно прост, доступен каждому практическому врачу. В то же время с его помощью можно получить немало полезной информации, что будет способствовать постановке правильного диагноза, а следовательно, назначению адекватного лечения и проведению необходимых профилактических мероприятий. Например, невральная амиотрофия Шарко - Мари и миотоническая дистрофия (наследственные болезни нервно-мышечной системы) имеют сходную клиническую картину (атрофии дистальной группы мышц с последующим парезом). Однако невральная амиотрофия Шарко - Мари наследуется по аутосомно-рецессивному типу, а миотоническая дистрофия - по аутосомно-доминантному. Установление типа наследования является решающим в постановке диагноза, от которого зависит прогноз для больного (болезнь Шарко - Мари приводит к инвалидности в течение 5-6 лет; миотоническая дистрофия течет более доброкачественно и работоспособность сохраняется длительное время), прогноз здоровья потомства этих больных и их лечение.
Одной из основных задач клинико-генеалогического метода является установление наследственного характера болезни. Если в семье встречается один и тот же признак или болезнь несколько раз, то в этом случае можно думать о наследственной природе или семейном характере болезни. Для установления наследственного характера патологии требуется большая тщательность сбора сведений о родственниках больного, полное клиническое и специальное лабораторное и инструментальное обследование определенного круга лиц в родословной (см. ниже). Чтобы не сделать ошибочного заключения о наследственном характере болезни там, где его нет, следует помнить о существовании уже упоминавшихся фенокопий наследственных болезней. Например, микроцефалия в сочетании с умственной отсталостью может явиться следствием редкой моногенной рецессивной мутации. В то же время некоторые препараты, принимаемые матерью во время беременности, или рентгеновское облучение плода могут вызвать аналогичные дефекты и представлять собой фенокопию генетически обусловленного заболевания. Подчас обнаруживаются случаи семейных заболеваний, обусловленных однотипным воздействием различных вредных факторов окружающей среды. "Накопление" болезни в родословной может наблюдаться, например, при семейном характере некоторых профессий. Одни и те же профессиональные вредности могут вызывать сходные заболевания у членов одной семьи. Если исключается действие сходных внешних факторов, можно предположить наследственный характер болезни. После того как обнаружен наследственный характер болезни, клинико-генеалогический метод позволяет установить тип наследования болезни: аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный доминантный или рецессивный. В случаях таких распространенных заболеваний с наследственной предрасположенностью, как артериальная гипертония, сахарный диабет, язвенная болезнь желудка и другие, с помощью клинико-генеалогического метода можно выявить роль наследственности в происхождении патологии.
Клинико-генеалогический метод имеет большое значение в клинической медицине в плане своевременной диагностики наследственных болезней. Некоторые болезни имеют типичную, легко выявляемую клиническую картину, относительно часто встречаются и поэтому легко диагностируются (например, гемофилия, полидактилия, дальтонизм). Другие наследственные болезни встречаются редко, а для некоторых имеются лишь единичные описания в мировой литературе. Распознать такие болезни практическому врачу трудно. Генеалогический метод дает возможность определить тип наследования болезни и тем самым нередко уточнить ее форму, поскольку для каждой наследственной патологии характерна передача по определенному типу. Особенности передачи наследственных болезней, устанавливаемые с помощью этого метода, позволяют правильно подойти к анализу ранних клинических симптомов, выявляемых у некоторых членов изучаемой семьи, и имеют дифференциально-диагностическое значение. Так, затруднена диагностика основных форм миопатии в начальных стадиях: псевдогипертрофической, ювенильной и плече-лопаточно-лицевой. Изучение генеалогических данных может помочь правильно оценить клинические симптомы болезни, определить ее форму, поскольку для псевдогипертрофической формы характерен сцепленный с полом тип наследования, для ювенильной - аутосомно-доминантный. С этих позиций данные генеалогического метода имеют большое значение для современной диагностики наследственных болезней - до развития выраженных стадий заболевания.
Генеалогический метод дает возможность определить круг лиц в родословной, нуждающихся в детальных исследованиях для выявления гетерозиготного носительства мутантного гена, что особенно важно для медико-генетического консультирования лиц с аутосомно-рецессивными и Х-сцепленными болезнями. Анализ родословной позволяет выявить родственников, нуждающихся в дополнительном обследовании.
В клинической медицине клинико-генеалогический метод служит и основой для изучения гетерогенности (многообразия) наследственных болезней. Принцип генетической гетерогенности состоит в том, что сходные по клиническим данным болезни могут быть вызваны мутациями в разных генах. Примером генетической гетерогенности является врожденная катаракта, которая может быть обусловлена аутосомно-рецессивным, аутосомно-доминантным генами и рецессивным геном, локализованным в хромосоме X. При этом клинические проявления будут совершенно идентичными. Используя клинико-генеалогический метод, можно определить характер наследования в конкретной семье. Генетическая гетерогенность выявлена для многих моногенных болезней.
Генеалогический метод широко используется при медико-генетическом консультировании, в частности для определения прогноза потомства в семьях, где есть или предполагается появление больного с наследственной патологией (см. ниже).
Применение клинико-генеалогического метода в клинической медицине для решения указанных выше вопросов можно рекомендовать в некоторых конкретных ситуациях, когда у больного и его родственников выявляются:
- известное моногенное заболевание;
- болезни с наследственной предрасположенностью (сахарный диабет, гипертоническая болезнь, шизофрения и др.);
- аналогичные заболевания или симптомы у нескольких членов семьи;
- хронические прогрессирующие заболевания неясного происхождения, не поддающиеся обычной терапии. Это может указывать, например, на иммунодефицитные состояния, муковисцидоз и т. д. Так, если у ребенка в раннем детском возрасте развивается острая пневмония, которая затем повторяется, становится затяжной, хронической и не поддается лечению, можно предположить муковисцидоз - наследственную патологию эндокринных желез;
- непереносимость некоторых пищевых продуктов (например, молока при галактоземии, белка злаков при целиакии и т. д.);
- извращенная реакция на действие лекарственных веществ. Например, появление гемолиза эритроцитов при лечении сульфаниламидами может быть вызвано генетически обусловленной недостаточностью глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ);
- различные аллергические заболевания в семье (например, аллергозы встречаются при некоторых патологиях обмена веществ);
- кровное родство родителей больного ребенка;
- отягощенный акушерский анамнез (бесплодие, выкидыши, мертворождения, ранняя детская смертность, воздействие вредных факторов в период беременности);
- врожденные пороки развития, особенно множественные.
В генеалогическом методе условно можно выделить два этапа: составление родословной и генеалогический анализ.
МЕТОДИКА СОСТАВЛЕНИЯ РОДОСЛОВНОЙ
Сбор генеалогической информации о наличии среди родственников больного тех или иных заболеваний может проводиться разными методами: опрос, очное и заочное анкетирование, личное обследование членов семьи. Основные трудности при применении клинико-генеалогического метода обычно бывают связаны с недостаточным объемом получаемой информации или ее искажением. Это обусловлено тем, что многие люди не знают состояние здоровья и причин смерти даже ближайших родственников и тем более родственников по линии мужа или жены. Естественно, чем больше членов семьи будет непосредственно опрошено, тем выше шансы на получение более достоверных и полных сведений. Необходимо объяснить больному цель тщательного опроса.
Иногда искажение сведений о родственниках жены (мужа) возникает при желании переложить на них "вину" за наследственную болезнь у ребенка. В таких случаях целесообразно опросить каждого из супругов отдельно, соблюдая тайну результатов опроса. Подчас больные сознательно скрывают сведения о наследственных болезнях, особенно таких, как нарушения психики, интеллекта, врожденные пороки развития, а также случаи тяжелых наследственных болезней у детей.
Ограниченность информации, получаемой при сборе сведений со слов, связана с особенностями самого метода, но в значительной степени зависит от умения провести опрос пациентов и установить с ними соответствующий психологический контакт, что особенно важно в работе семейного врача.
Метод анкетирования - второй прием, применяемый для сбора генеалогических данных. Он может дополнять первый или использоваться самостоятельно. Иногда его результаты оказываются более достоверными, чем метода непосредственного опроса пациента. Анкеты заполняют больные, его родственники или лечащий врач. Анкеты содержат различный перечень вопросов в зависимости от целей проводимого обследования. Метод заочного анкетирования применяется в том случае, если родственники больного проживают, например, в другом городе и недоступны для обследования, а получение медицинской информации от них крайне необходимо.
При составлении родословной с целью постановки или же подтверждения диагноза можно использовать выписки из историй болезни, карты новорожденного, протоколы патолого-анатомических исследований.
Молекулярные основы наследственной патологии Ферментопатии Лечение наследственных болезней Заместительная терапия Витаминотерапия Индукция и ингибиция метаболизма Хирургическое лечение Диетотерапия Эффективность лечения мультифакториальных болезней в зависимости от степени наследственного отягощения у больных Разрабатываемые методы лечения Профилактика врожденной патологии у женщин из групп повышенного риска Клиническая фармакогенетика Наследственные дефекты ферментных систем, выявляемые при применении лекарств Атипичные реакции на лекарства при наследственных болезнях обмена веществ Наследственная обусловленность кинетики и метаболизма лекарств Генетические основы тестирования индивидуальной чувствительности к лекарствам Медико-генетическое консультирование Задачи и показания для проведения консультации Принципы консультирования Этапы консультирования Пренатальная диагностика врожденных пороков развития и наследственных болезней Проблемы медико-психологической реабилитации больных с врожденными болезнями и членов их семей Умственная отсталость Дефекты зрения и слуха Аномалии опорно-двигательного аппарата Приложения Блок информации N 1 - ишемическая болезнь сердца Блок информации N 2 - сахарный диабет Блок информации N 3 - язвенная болезнь Блок информации N 4 - врожденные пороки развития на примере расщелины губы и/или неба Литература [показать]
|
Генеалогический метод исследования наследственности человека
В настоящее время медицинская генетика располагает огромным количеством методов исследования, позволяющих решать подавляющее большинство практических и теоретических вопросов. Ряд из этих методов имеет уже большую историю (генеалогический, цитологический, близнецовый), другие возникли недавно, но получили неоценимое значение, как для теории, так и для практики (иммунологический, ДНК-зондовая диагностика и т.д.)
Изучение генетики человека связано с рядом особенностей и объективных трудностей:
позднее половое созревание и редкая смена поколений;
малое количество потомков;
невозможность экспериментирования;
невозможность создания одинаковых условий жизни.
Цитогенетические методы исследования генетики человека основаны на исследовании человеческого кариотипа (хромосомный набор, совокупность признаков хромосом в клетках тела).
Этапы исследования клеток человека на искусственных питательных средах; проведение специальных манипуляций, вследствие чего хромосомы «рассыпаются» и лежат свободно; окрашивание хромосом; изучение хромосом под микроскопом и фотографирование; вырезание отдельных хромосом и построение детального изображения хромосомного набора.
В 70-е годы были разработаны методы дифференциального окрашивания хромосом человека, которые позволили выявлять геномные (например, болезнь Дауна) и хромосомные (например, синдром «кошачьего крика») мутации.
Существуют молекулярно-цитогенетические методы, которые основаны на методе FISH, с помощью которого можно определять локализацию генов в хромосомах и все хромосомные отклонения от нормы.
Биохимические методы
Практические все биохимические реакции, протекающие в человеческом организме и в конечном итоге составляющие его обмен веществ, регулируются ферментами. Биохимические методы изучения генетики человека основаны на изучении активности ферментных систем. Активность оценивают или по активности самого фермента, или по количеству конечных продуктов реакции, которую контролирует данный фермент
Применяются разнообразные методы изучения, среди них хроматографические, флюорометрические, радиоиммунологические и др. Изучения активности ферментных систем позволяет выявлять генные мутации, которые являются причинами болезней обмена веществ, например, фенилкетонурии, серповидно-клеточной анемии.
С помощью биохимических методов можно выявлять носителей патологического генов таких болезней как, например фенилкетонурия, сахарный диабет и др.
Близнецовый метод
В 1876 году Ф.Гальтоном был введен в медицинскую практику близнецовый метод изучения генетики человека. Он позволяет определить роль генотипа (совокупность наследственных свойств) и окружающей среды в проявлении признаков болезни.
Различают моно- и дизиготных близнецов.
Монозиготные (однояйцевые) близнецы развиваются из одной оплодотворенной яйцеклетки. Они имеют одинаковый генотип, но могут отличаться по фенотипу (совокупность внешних и внутренних признаков и свойств, сформировавшихся на базе генотипа в процессе развития) что обусловлено воздействием факторов внешней среды.
Монозиготные близнецы имеют большую степень сходства по признакам, которые определяются преимущественно генотипом: они всегда однополы, имеют одинаковые группы крови, один цвет глаз, однотипные узоры на пальцах и ладонях и др.
Дизиготные (двуяйцевые) близнецы развиваются после оплодотворения одновременно созревших яйцеклеток. Они имеют разный генотип, и их фенотипические отличия обусловлены как генотипом, так и факторами внешней среды.
Таким образом, фенотипические признаки и используются для определения зиготности близнецов.
Процент сходства близнецов по изучаемому признаку называется конкордантностью, а процент различия дискордантностью.
Для оценки роли наследственности и окружабщей среды в развитии болезни используется формула Хольцингера:
КМБ (%) – КДБ (%) / 100% - КДБ(%), где Н – доля наследственности, КМБ – конкордантность монозиготных близнецов, КДБ – конкордантность дизиготных близнецов.
Если результат расчетов по формуле Хольцингера приближается к единице, то основная роль развития болезни принадлежит наследственности. И наоборот, если результат стремится к нулю, большую роль сыграли факторы окружающей среды.
Популяционно-статистический метод изучения генетики человека основан на использовании математического выражения закона Харди-Вейнберга.
Нужно взять на р. Частоту встречаемости в популяции доминантного гена, за q частоту встречаемости рецессивного гена, за p2 частоту доминантных гомозигот, за 2pq частоту рецессивных гомозигот, за 2pq частоту гетерозигот.
Сумма частот всех генотипов должна быть принята за 1 (100%): p2 +2pq+q2=1(100%).
Метод позволяет определять частоту генов в генотипе в больших (свыше 4,5 тыс.) популяциях.
Современные методы пренатальной диагностики наследственных и врожденных заболеваний.
Пренатальная диагностика – это дородовое определение врожденной или наследственной патологии у плода.
С организационной точки зрения все беременные (без специальных показаний) должны обследоваться для исключения наследственной патологии просеивающими методами (УЗИ, биохимическое исследование сыворотки беременных).
Показаниями для пренатальной диагностики являются:
наличие в семье точно установленного наследственного заболевания;
возраст матери старше 35 лет, отца старше 45 лет;
наличие у матери Х-сцепленного рецессивного патологического гена;
беременные, имеющие в анамнезе спонтанные аборты, мертворождения неясного генеза, детей с множественными врожденными пороками развития и с хромосомной патологией;
наличие структурных перестроек хромосом у одного из родителей;
гетерозиготность обоих родителей при аутосомно-рецессивных заболевания.
В пренатальной диагностике используются инвазивные и неинвазивные методы.
Неинвазивные методы включают:
ультразвуковое исследование плода по меньшей мере два раза (12-14 недель и 20-21 недели беременности). С помощью УЗИ диагностируются пороки развития конечностей, дефекты невральной трубки, гидро- и микроцефалия, пороки сердца, аномалии почек;
биохимические методы включают определение уровня альфа-фетопротеина, хорионического гонадотропина, несвязанного эстрадиола в сыворотке крови беременных. Эти методы выявляют пороки развития, многоплодную беременность, внутриутробную гибель плода, маловодие, угрозу прерывания, хромосомные заболевания плода и другие патологические состояния. Оптимальные сроки исследования – 17-20 недель беременности.
Инвазивная пренатальная (дородовая) диагностика включает методы, при которых для исследования получают клетки плода или окружающих его тканей и структур. Такие методы сопровождаются повышенным риском невынашивания беременности и антенатальной гибели плода. Вероятность преждевременного прерывания беременности колеблется в зависимости от вида метода исследования и составляет от 1 до 6%. Поэтому инвазивная диагностика может использоваться в тех случаях, когда риск рождения больного ребёнка превышает возможности осложнений периода беременности.
Методы исследования тканей плода постоянно совершенствуются, чтобы обеспечить наиболее раннее, безопасное и достоверное выявление наследственных заболеваний. В последние годы наиболее широко распространены следующие способы инвазивной диагностики:
амниоцентез – процедура получения амниотической жидкости путём пункции амниотического мешка через переднюю брюшную стенку под контролем ультразвука. Проводится в сроки беременности 15-18 недель. Полученную амниотическую жидкость подвергают последующему биохимическому исследованию, а клетки плода служат материалом для цитогенетического исследования или ДНК-диагностики. Можно диагностировать все хромосомные болезни и ряд генных заболеваний. При проведении амниоцентеза возможны осложнения (гибель плода, инфицирование полости матки).
хорионбиопсия проводится на 9-13 недели беременности. Исследуемым материалом являются нативные клетки и структура клеток тканей хориона. Клетки ворсин хориона несут такую же информацию, как и клетки плода. Могут быть выявлены хромосомные нарушения, более 100 болезней обмена веществ: галактоземия, гликогенозы II, III, IV типа, болезнь Тея-Сакса и др.Примерно в 2,5-3% случаев биопсия хориона провоцирует самопроизвольное прерывание беременности, гибель плода или внутриматочное инфицирование.
кордоцентез. Метод заключается во взятии крови из пуповины плода под ультразвуковым контролем. Проводится в сроки 20-23 недели, может быть использован и для внутриутробного лечения – введения лекарственных веществ. Риск осложнений составляет около 2%. Применяют данный метод для выявления хромосомных заболеваний, иммунодефицитов, инфекций, ДНК-диагностики генных болезней.
фетоскопия и фетоамниография. Фетоскопия включает введение в полость матки специального прибора – фетоскопа, созданного на основе волоконно-оптической техники. Помимо выявления видимых снаружи дефектов плода, при данных исследованиях возможно осуществить биопсию кожи или печени плода. Исследование обычно используется только для диагностики тяжелых кожных заболеваний (ихтиоз, буллезный эпидермолиз). Проводится во втором триместре беременности (18-24 нед.), характеризуется 6-8 %-ным риском осложнений.
Массовые просеивающие программы.
Программа ранней диагностики наследственных болезней подразумевает массовое просеивание (скрининг) наследственных болезней обмена у всех новорожденных.
В европейских странах массовый скрининг проводится для доклинического выявления фенилкетонурии, гипотиреоза, врожденной гиперплазии коры надпочечников, галактоземии и муковисцидоза.
В Беларуссии массовое просеивание новорожденных на фенилкетонурию и гипотиреоз осуществляется почти повсеместно.
Генеалогический мет од является одним из первых научных методов исследования в медицинской генетике. Это метод изучения родословных, с помощью которого прослеживается распределение болезни (признака) в семье или роду с указанием типа родственных связей между членами родословной. Метод часто называют клинико-генеалогическим, поскольку речь идёт об изучении патологических признаков (болезней) в семье с привлечением приёмов клинического обследования.
В настоящее время метод позволяет решать ряд немаловажных вопросов и в частности:
устанавливать является ли данный признак или заболевание единичным в семье или имеются несколько случаев данной патологии;
выделять лиц подозрительных в отношении данного заболевания и составлять план их обследования для уточнения диагноза;
определять тип наследования и выяснять, по какой линии, материнской или отцовской, идёт передача заболевания;
выявлять лиц, нуждающихся в медико-генетическом консультировании, определять клинический прогноз для пробанда и его больных родственников с учётом особенностей заболевания и его генетической характеристики;
разрабатывать план лечения и профилактики с учётом индивидуальных и семейных особенностей заболевания;
прогнозировать вероятность проявления наследственной патологии в последующих поколениях в зависимости от типа наследования.
При клинико-генеалогическом методе выделяют два последовательных этапа:
составление родословной и её графическое изображение;
генетический анализ полученных данных.
Сбор сведений о семье начинается с пробанда – обследуемого человека, больного или здорового. При составлении родословной обычно пользуются условными обозначениями. Для составления родословной необходимы сведения не менее чем о 3-4-х поколениях семьи пробанда. Необходимо собирать сведения, касающиеся не только наличия конкретного заболевания или патологического признака, но и информацию обо всех случаях заболеваний, встречающихся среди членов семьи, спонтанных абортах, мертворождениях и ранней детской смертности.
Графическое изображение родословной (ввел Г. Юст в 1931году, используется в настоящее время):
Обследуемые братья и сёстры (сибсы), их жёны и мужья одного поколения располагаются в одном ряду слева направо в порядке рождения и обозначаются арабскими цифрами;
поколения обозначаются римскими цифрами;
любая родословная сопровождается пояснениями (легенда), где указываются данные о том или ином родственнике, который подлежит обследованию; возраст; начало и характер течения заболевания у пораженного; причину смерти и возраст на момент смерти члена родословной; описание методов диагностики заболеваний и др. сведения.
Генеалогический анализ родословной включает:
Установление наследственного характера признака. Если исключить действие сходных внешних факторов (фенокопий), то можно думать о наследственном характере заболевания.
Установление типа наследования. Для этого используют принципы генетического анализа и различные статистические методы обработки данных, полученных из родословной.
Выделяют пять основных типов наследования. Критерии аутосомно-доминатного, аутосомно-рецессивного, Х-сцепленного доминатного, Х-сцепленного рецессивного типов наследования мы с Вами разбирали (см. лекция №3).
Мультифакториальное наследование, критерии:
Методы исследования медицинской генетики
Контрольная работа >> Биология... исследований – плодовая мушка дрозофила. Исследования проводятся с 1902г. 1. ГЕНЕАЛОГИЧЕСКИЙ МЕТОД ... способностей человека . В настоящее время генеалогический метод ... МЕТОД С помощью биохимических методов определяется многочисленная группа наследственных ...
Современные методики исследования психогенетики человека
Реферат >> Биология... наследственности и среды в формировании психических и психофизиологических свойств человека занимается психогенетика. Целью исследований ... метода и препятствием в установлении генофонда популяции. 2.3. Генеалогический метод Генеалогический метод ...
высокая частота в популяции (сахарный диабет, артериальная гипертензия и т.д.);
несоответствие законам Г.Менделя;
существование различных клинических форм;
чем реже болезнь встречается в популяции, тем выше риск для родственников больного заболеть этой же формой;
чем сильнее выражена болезнь у пробанда, тем выше риск болезни для его родственников;
риск заболеть у родственников тем выше, чем выше степень родства с больным членом семьи (выше число общих генов);
Реферат >> ПсихологияМожет иметь и негенетическое происхождение. Генеалогический метод - исследование сходства между родственниками в разных поколениях... детьми одного человека . Метод широко используется при изучении наследственных причин ряда заболеваний. Исследование близнецов...
Генеалогический метод
генеалогический наследственный заболевание
Генеалогический метод состоит в изучении родословных на основе менделеевских законов наследования и помогает установить характер наследования признака (доминантный или рецессивный).
Так устанавливают наследование индивидуальных особенностей человека: черт лица, роста, группы крови, умственного и психического склада, а также некоторых заболеваний. Например, при изучении родословной королевской династии Габсбургов в нескольких поколениях прослеживаются выпяченная нижняя губа и нос с горбинкой.
Этим методом выявлены вредные последствия близкородственных браков, которые особенно проявляются при гомозиготности по одному и тому же неблагоприятному рецессивному аллелю. В родственных браках вероятность рождения детей с наследственными болезнями и ранняя детская смертность в десятки и даже сотни раз выше средней.
Генеалогический метод чаще других используется в генетике психических болезней. Его сущность состоит в прослеживании в родословных проявлений патологических признаков с помощью приемов клинического обследования с указанием типа родственных связей между членами семей.
Этот метод используется для установления типа наследования болезни или отдельного признака, определения местоположения генов на хромосомах, оценки риска проявления психической патологии при медико-генетическом консультировании. В генеалогическом методе можно выделить 2 этапа - этап составления родословных и этап использования генеалогических данных для генетического анализа.
Составление родословной начинают с человека, который был обследован первым, его называют пробандом. Обычно это бывает больной или индивид, у которого есть проявления изучаемого признака (но это не обязательно). Родословная должна содержать краткие сведения о каждом члене семьи с указанием его родства по отношению к пробанду. Родословную представляют графически, используя стандартные обозначения, как это показано на рис. 16. Поколения указывают римскими цифрами сверху вниз и ставят их слева от родословной. Арабскими цифрами обозначают индивидов одного поколения последовательно слева направо, при этом братья и сестры или сибсы, как их называют в генетике, располагаются в порядке даты их рождения. Все члены родословной одного поколения располагаются строго в один ряд и имеют свой шифр (например, III-2).
По данным о проявлении заболевания или какого-то изучаемого свойства у членов родословной с помощью специальных методов генетико-математического анализа решается задача установления наследственного характера заболевания. Если установлено, что изучаемая патология имеет генетическую природу, то на следующем этапе решается задача установления типа наследования. Следует обратить внимание на то, что тип наследования устанавливается не по одной, а по группе родословных. Подробное описание родословной имеет значение для оценки риска проявления патологии у конкретного члена той или иной семьи, т.е. при проведении медико-генетического консультирования.
При изучении различий между индивидами по любому признаку возникает вопрос о причинных факторах таких различий. Поэтому в генетике психических заболеваний широко используется метод оценки соотносительного вклада генетических и средовых факторов в межиндивидуальные различия по подверженности тому или иному заболеванию. Этот метод основан на предположении, что фенотипическое (наблюдаемое) значение признака у каждого индивида является результатом влияния генотипа индивида и тех условий среды, в которых происходит его развитие. Однако у конкретного человека определить это практически невозможно. Поэтому вводятся соответствующие обобщенные показатели для всех людей, позволяющие затем в среднем определить соотношение генетического и средового влияния на отдельного индивида.
Изучение генеалогическим методом семей лиц, страдающих психическими болезнями, убедительно показало накопление в них случаев психозов и аномалий личности. Увеличение частоты случаев болезни среди близких родственников было установлено для больных шизофренией, маниакально-депрессивным психозом, эпилепсией, некоторыми формами олигофрении.
При генетическом анализе важно учитывать клиническую форму заболевания. В частности, частота шизофрении среди родственников во многом зависит от клинической формы болезни, которой страдает пробанд.
Величины риска, приведенные в таблицах, позволяют врачу ориентироваться в вопросах наследования заболевания. Например, наличие в семье (кроме самого пробанда) еще одного больного родственника повышает риск для остальных членов семьи, причем не только тогда, когда больны оба или один родитель, но и тогда, когда больны другие родственники (сибсы, тети, дяди и др.).
Таким образом, близкие родственники больных психическим заболеванием имеют повышенный риск по аналогичной болезни. Практически можно выделять: а) группы повышенного риска - дети, один из родителей которых болен психическим заболеванием, а также сибсы (братья, сестры), дизиготные близнецы и родители больных; б) группы наивысшего риска - дети двух больных родителей и монозиготные близнецы, один из которых заболел. Ранняя диагностика, своевременная квалифицированная психиатрическая помощь составляют суть профилактических мероприятий в отношении этого контингента.
Результаты клинико-генетических исследований составляют основу медико-генетического консультирования в психиатрии. Медико-генетическое консультирование схематически можно свести к следующим этапам:
установление правильного диагноза пробанду;
составление генеалогии и изучение психического состояния родственников (для правильной диагностической оценки в этом случае особенно важна полнота сведений о психическом состоянии членов семьи);
определение риска по заболеванию на основании данных;
оценка степени риска в понятиях «высокий - низкий». Данные о риске сообщают в форме, соответствующей потребностям, намерениям и психическому состоянию консультирующегося лица. Врач должен не только сообщить степень риска, но и помочь правильно оценить полученные сведения, взвесив все «за» и «против». Следует также устранить у консультирующегося чувство вины за передачу предрасположения к болезни;
формирование плана действия. Врач помогает в выборе того или иного решения (иметь детей или отказаться от деторождения могут только сами супруги);
катамнез.
Наблюдение за семьей, обратившейся за консультацией, может дать врачу новые сведения, способствующие уточнению степени риска.
Заключение
Хотя человек является сложным объектом для генетических исследований, поскольку у человека большое число генов, высока степень их гетерозиготности, невозможны направленные скрещивания и др., все же наследственность человека подчиняется универсальным для всего органического мира законам, а особенности наследственности каждого человека можно выявить, используя генеалогический метод генетического анализа.
У человека имеют место различные типы наследования.
Наследование признаков человека подчиняется общим генетическим закономерностям.
Чтобы выявить тип наследования у человека необходим специальный метод - генеалогический метод.
Список литературы
- 1. Айала Дж., Кайгер Ф. Современная генетика. - М.: Мир, 1987.
- 2. Бочков Ф.П. Генетика человека. - М.: Просвещение, 1990.
- 3. Спицын И.П. Практикум по генетике человека. - Тамбов, 1999.
- 4. Фогель А., Мотульски К. Генетика человека - М.: Мир, 1990. - Т. 1-3.
У человека как объекта генетического исследования почти нет никаких преимуществ перед другими объектами.
Напротив, много препятствий, затрудняющих изучение его генетики: 1) невозможность произвольного скрещивания в эксперименте; 2) позднее наступление половой зрелости; 3) малое число потомков в каждой семье; 4) невозможность уравнивать условия жизни для потомства; 5) отсутствие точной регистрации проявления наследственных свойств в семьях и отсутствие гомозиготных линий; 6) большое число хромосом; 7) и самым главным затруднением изучения генетики человека в капиталистическом обществе является социальное неравенство, что затрудняет реализацию наследственных потенций человека.
Несмотря на указанные затруднения, генетика разработала некоторые методы, которые позволяют шаг за шагом изучать наследственность и наследование у человека. Существует несколько методов исследования: генеалогический, цитогенетический, близнецовый, онтогенетический и популяционный.
Следует иметь в виду, что любой признак, независимо от того, является ли он признаком дикого типа, т. е. относится к норме, или связан с каким-либо заболеванием, может служить моделью для изучения наследственности. Оградить человека от наследственных болезней или поражения его наследственности так же важно, как и выяснить наследование нормы. В настоящее время генетические методы разработаны главным образом в отношении морфологических признаков, которые генетически определяются достаточно четко (брахидактилия, альбинизм, дальтонизм, пятнистость кожи и волос и т. д.).
Генетическое исследование психических свойств все еще остается проблематичным, так как для них не найдены элементарные критерии признака в генетическом смысле. Почти все признаки Психической и творческой деятельности человека настолько комплексны и сложны, а также в сильной степени обусловлены внешними, в том числе и социальными, факторами, что генетический анализ этих свойств пока трудно осуществим, хотя наследственная их обусловленность не вызывает сомнения.
Можно сказать, что значительное большинство признаков, характеризующих вид Homo sapiens, может изучаться как количественные и сложные физиологические признаки, т. е. признаки, не проявляющие дискретного характера в онтогенезе. Эти признаки контролируются системой генотипа (полигенно). И пока эта система не разгадана хотя бы на примере просто организованных организмов, проблема признаков поведения остается малодоступной для генетического анализа. Напротив, мутантные признаки, выходящие за границы характеристики видовых признаков, служат хорошими генетическими моделями изучения наследственности и наследования в норме.
На дискретные мутантные признаки нельзя смотреть как на признаки только патологические, якобы не имеющие приспособительного значения. Возможно, что само появление человека с развитыми полушариями коры головного мозга, вертикальным положением тела, дискретной речевой сигнализацией является следствием крупных мутаций. В пользу этого свидетельствует очень
короткий промежуток времени эволюции человека, за который мелкие мутации вряд ли могли накопиться в таком количестве и дать такой значительный эволюционный эффект. Разумный человек для природы столь же «необычен», как домашняя курица, несущая 365 яиц в год вместо 10-15, или рекордистка-корова, дающая 16 тыс. кг молока в год вместо 600-700 кг.
Разделение признаков на нормальные и мутантные применительно к человеку и животным необходимо для познания эволюции человека и патологических явлений.
Совокупность видовых признаков человека и животных определяется системой генотипа, сложившейся под влиянием всех факторов отбора в процессе эволюции. Мутации, пребывающие в гетерозиготном состоянии у человека, по-видимому, так же необходимы, как и у животных, для поддержания их в популяции.
Самым опасным в разработке научных методов исследования животных и человека, особенно его способностей, является антропоморфический момент, т. е. выдача желаемого за действительность.
Генеалогический метод
Анализ наследования человека на основе составления родословной - генеалогии был предложен Ф. Гальтоном.
Генеалогический метод представляет собой изучение наследования свойств человека по родословным (педигри). Данный метод применим, если известны прямые родственники - предки обладателя наследственного признака (пробанда) по материнской и отцовской линиям в ряду поколений и имеется достаточное число потомков в каждом поколении, или в том случае, когда имеются данные по достаточному числу разных семей, позволяющему выявить сходство родословных. Данные по совокупности сходных родословных подвергают статистической обработке.
Получившая наибольшее распространение система обозначения родословных человека была предложена Г. Юстом в 1931 г.
На основе большого числа проанализированных семей составляют родословные и производят математические расчеты соответственно типу наследования того или иного признака - доминантному или рецессивному, часто и не часто встречающейся мутации, сцепленному или не сцепленному с полом и т. д. Здесь мы не будем касаться приложения математического метода к данному анализу, отметим только, что весь этот формальный анализ основан на элементарных генетических закономерностях наследования.
Схемы родословной наследования дохминантного аутосомного гена, определяющего какой-либо признак, например заболевание (хондродистрофическая карликовость, буллезный эпидермолиз - свойство кожи образовывать большие пузыри при небольших травмах, ретинобластома и т. д.), или морфологический недостаток, например короткопалость (брахидактилия - отсутствие двух дистальных фаланг в пальцах).

Наследование признаков, определяемых рецессивными генами (рецессивное наследование), анализируется несколько сложнее, при составлении схем родословных.
Например, двоих в семье, появление двоих больных детей равно произведению вероятностей, т. е. 0,25 X 0,25, т. е. 6,25%.
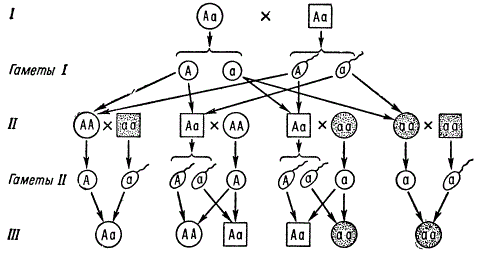
Часто встречающиеся рецессивные аутосомные гены при условии, если носители их (аа) способны вступать в брак и давать потомство, будут находиться в высокой концентрации в популяции. В таком случае становятся очень вероятными браки аа X Аа, в потомстве от которых наследование данного признака будет имитировать наследование по доминантному типу 1:1. Однако, зная тип наследования и проявления тех и других генов даже в случае малочисленных семей, но при достаточном числе таких семей, можно установить истинный характер наследования.
Наследование генов, полностью сцепленных с полом, т. е. находящихся в негомологичных сегментах, и частично сцепленных с полом - локализованных в гомологичных сегментах X- и Y-xpoмосом, подчиняется установленным для половых хромосом закономерностям. Для доминантных и рецессивных генов это наследование будет определяться по-разному в зависимости от того, где данный ген локализован - в гомологичном или негомологичном сегменте X- и Y-хромосомы и каким образом он передается. Так, доминантный ген, вызывающий перепончатость пальцев, находящийся в негомологичном сегменте Y-хромосомы, наследуется от отцов и проявляется только у мужчин.
Для частично сцепленных с полом доминантных генов, находящихся в гомологичных сегментах половых хромосом, анализ несколько более затруднен, но также возможен. Примером сцепленного с полом наследования рецессивного признака является наследование гемофилии. В передаче этого признака в поколениях имеется прерывность; пораженные мужчины являются потомками здоровых матерей, которые были гетерозиготами по данному гену; больные гемофилией женщины могут быть потомками больного отца и больной или здоровой матери.

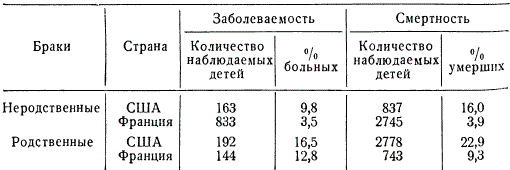
У человека найдено около 50 сцепленных с полом рецессивных генов. Интересно, что около половины из них обусловливают заболевание глаз. Уже с давних времен было известно, что степень передачи наследственных признаков в родственных (инбридинг) и неродственных браках (аутбридинг) различна. После того, как генетика установила закономерности более частого проявления рецессивных генов при инбридинге, нет необходимости пространно доказывать вред родственных браков. Чем выше коэффициент инбридинга, тем больше вероятность появления наследственных болезней в поколениях. В разных странах среди разных народов и классов общества, а также в разные эпохи родственные браки (между двоюродными, троюродными братьями и сестрами) встречаются с разной частотой. Так, например, в деревнях на островах Фиджи количество родственных браков достигает 29,7%, в некоторых кастах Индии - 12,9, в Японии (Нагасаки) - 5,03, в Голландии - 0,13-0,159, в Португалии- 1,40, в США (Балтимора) - 0,05%, и т. д. Процент родственных браков колеблется в отдельных районах одной и той же страны в зависимости от уклада жизни.
Вредность родственных браков мало заметна в отдельных родословных, но при сравнительном статистическом анализе болезней и смертностей она выступает с полной очевидностью.
Яркий пример выявления рецессивного гена при родственном браке.

В этой родословной родство поддерживается через бракосочетание сибсов (братья - сестры) разной степени родства. От двух родственных браков (четвероюродные сибсы) появилось в одной семье 4 ребенка из 8, а в другой - 2 из 5, страдающих наследственной амавротической идиотией. К. Штерн предполагает, что один из двух общих предков этих линий передал данный рецессивный ген через три поколения каждому из четырех родителей.
Иногда заболевание и смертность детей от родственных браков превышают на 20-30% таковые от неродственных браков. Очевидно, что причина рассматриваемого явления генетическая, а именно: большая вероятность проявления наследственных заболеваний и смертности в результате гомозиготизации рецессивных генов, определяющих физиологические недостаточности и смертность (летальные и полулетальные гены).
Итак, генеалогический метод является весьма ценным методом, однако его значение в исследованиях тем больше, чем точнее и глубже составлены родословные. По мере роста цивилизации и более точной регистрации родословных роль этого метода в генетике человека будет возрастать.
Близнецовый метод
Близнецами называют потомство, состоящее из одновременно родившихся особей у одноплодных животных (человек, лошадь, крупный рогатый скот, овцы и др.).
Близнецы могут быть однояйцевыми и разнояйцевыми.
Идентичные, или однояйцевые, близнецы (ОБ) развиваются из одного яйца, оплодотворенного одним сперматозоидом, когда из зиготы вместо одного зародыша возникают два или более (полиэмбриония). В силу того, что митотическое деление зиготы дает два равнонаследственных бластомера, однояйцевые близнецы, сколько бы их ни развивалось, должны быть наследственно идентичны и одного пола. Это явление представляет собой пример бесполого, а точнее, вегетативного размножения животных.
Разнояйцевые близнецы (РБ) развиваются из одновременно овулировавших разных яйцеклеток, оплодотворенных разными сперматозоидами. И так как разные яйцеклетки и сперматозоиды могут нести различные комбинации генов, то разнояйцевые близнецы могут быть наследственно столь же разными, как и дети одной и той же супружеской пары, родившиеся в разное время. Разнояйцевые близнецы могут быть одного (РБо) или разного пола (РБр).
Чаще в литературе вместо термина «разнояйцевые близнецы» (РБ) употребляют термин «двуяйцевые близнецы» (ДБ), так как двойни встречаются чаще. Однако термин «разнояйцевые близнецы» лучше подчеркивает разницу между ОБ и РБ; однояйцевые близнецы также чаще рождаются двойнями.
Судя по данным, полученным на млекопитающих, для объяснения образования ОБ у человека может быть несколько гипотез:
- расхождение бластомеров при первом дроблении зиготы и раздельное развитие зародыша из этих бластомеров;
- разделение группы клеток на стадии бластоциста (до гаструляции);
- разделение зародышей на ранней стадии гаструляции. Наиболее вероятным путем предполагают второй.

Число близнецов в одних родах у человека колеблется: чаще всего встречаются двойни, реже тройни, еще реже - четверни, совсем редко - пятерни. По данным И. И. Канаева, за последние 150 лет в США установлено четыре случая родов пятерни, в Канаде - два случая. Факт рождения ОБ - пятерни девочек, доживших до взрослого состояния, - известен в семье канадского фермера Дионн (1934 г.). Рассчитано, что пятерни рождаются один раз на 54 700 816 родов, шестерни - на 4712 млн. родов, семерни известны только как исключение. В среднем частота рождения близнецов составляет 1% с колебаниями в пределах 0,5-1,5%. Близнецы менее жизнеспособные, и поэтому их количество при рождении меньше, чем при зачатии, а во взрослом состоянии меньше, чем при рождении.
Расчет частоты ОБ по отношению к РБ делается исходя из теоретического соотношения однополых и разнополых пар РБ при рождении близнецов: 25%♀♀ + 50%♀♂ + 25%♂♂ вычитание числа пар разного пола из общего числа всех пар одинакового пола (мужского и женского) даст разницу, составляющую число пар ОБ, которая в среднем колеблется от 21 до 33,4% всех близнецов.
Для использования близнецов в генетических исследованиях очень важно точно диагностировать тип ОБ и тип РБ. Диагностика производится на основании следующих критериев:
- ОБ обязательно одного пола, РБ могут быть как одного пола, так и разных полов;
- ОБ имеют, как правило, один общий хорион, РБ - разные хорионы;
- реципрокная трансплантация тканей у ОБ столь же успешна, как и автотрансплантация, у РБ она невозможна;
- наличие сходства (конкордантности) у ОБ и несходства (дискордантности) у РБ по многим признакам.
Для диагностики следует выбирать признаки, четко наследующиеся и менее всего подверженные изменению под влиянием факторов среды; к таким признакам относятся группы крови, пигментация глаз, кожи и волос, кожный рельеф (отпечатки кончиков пальцев, ладоней, ступней и др.). Если по одному-двум таким признакам выявлено различие близнецов, то они, как правило, являются РБ.
Все сомнительные случаи диагностики близнецов могут быть вызваны либо нарушением развития одного из партнеров ОБ, либо сходством родителей по ряду признаков. Однако последнее встречается чрезвычайно редко. Следует заметить, что нарушение развития одного из партнеров ОБ обычно объясняют неодинаковым действием факторов внутриутробной жизни и возникновением соматических мутаций на ранних стадиях эмбрионального развития, до закладки органов. Различного рода генные и хромосомные перестройки, моносомия и другие мутации, возникающие у одного из партнеров, способны вызвать значительные различия в фенотипе ОБ. Поэтому необходимо учитывать возможность соматических мутаций у ОБ в раннем эмбриогенезе.
Согласно обобщениям И. И. Канаева, изложенным в его превосходной монографии сущность близнецового метода в генетике сводится к следующим положениям:
1) пара ОБ имеет тождественную комбинацию, пара РБ - разные комбинации генотипов родителей;
2) для обоих партнеров одной пары ОБ внешняя среда может оказаться одинаковой, а для другой - разной. Если партнеры ОБ в течение жизни испытывают разное влияние, то это приведет к внутрипарному различию. Отсюда пары могут быть с внутрипарной одинаковой и внутрипарной разной средой.
Сравнение ОБ с одинаковой средой с ОБ с разной средой открывает возможность судить о роли влияния среды на внутрипарные различия близнецов в течение всей жизни. Сравнение ОБ с одинаковой средой и РБ с одинаковой средой позволяет выяснить роль наследственного фактора. Такого рода изучение проводят на большой выборке и обрабатывают статистически.
Исходя из разности генетического происхождения ОБ и РБ вытекает, что если по одним и тем же признакам нет различия у ОБ и есть таковые у РБ, то очевидно, что данные различия признаков у последних обусловлены наследственными факторами. Если же внутрипарные различия в тех же признаках встречаются у одного и другого типа близнецов, то очевидно, что они могут быть вызваны факторами среды. Из данных дискордантности у ОБ и РБ по ряду морфологических признаков, видно, что внутрипарное различие у РБ встречается во много раз чаще, чем у ОБ.

Представлены некоторые данные С. Рида относительно сравнительной частоты патологии у второго партнера в случае заболевания одного из близнецов.

В процентах показана частота конкордантности заболеваний у двух типов близнецов, из него видно, что если один партнер заболел одной из указанных болезней, то вероятность заболевания второго у ОБ значительно выше, чем у РБ. В. П. Эфроимсон, анализируя данные по частоте контордантных пар, совершенно правильно указывает, что высокая Наследственная предрасположенность ОБ к заболеваниям проявляется при наличии провоцирующего фактора; без него этот процент будет значительно ниже.
Близнецовый метод дает возможность с наибольшей точностью выяснить наследственную предрасположенность человека к ряду заболеваний и свойств. Другими методами очень трудно или почти невозможно исследовать многие инфекционные и опухолевые заболевания, воспаления кожи и различных органов, а также характеристики нормальной нервной деятельности человека.
При использовании близнецового метода приходится учитывать условия совместного и раздельного воспитания в жизни партнеров, социальные условия, в которых они находятся, и т. д. Тем не менее близнецовый метод позволяет наиболее точно определить, коэффициент наследуемости разных признаков, а также судить о гетерогенности популяции по изучаемым генам и вычленять роль среды в определении изменчивости изучаемых признаков.
Цитогенетический метод
Цитогенетическим методом в генетике человека обычно называют цитологический анализ кариотипа человека в норме и патологии.
Правильнее этот метод называть цитологическим, а не цитогенетическим, поскольку генетический анализ путем скрещивания у человека исключен, и носители хромосомных нарушений если выживают, то оказываются, как правило, бесплодными. Однако изредка в отношении некоторых хромосомных нарушений удается сочетать цитологический метод с генеалогическим и устанавливать связь фенотипического эффекта с определенным типом хромосомных изменений. В силу этих обстоятельств можно сохранить принятый в литературе термин «цитогенетический метод» в изучении генетики человека. В тех же случаях, где такого параллелизма исследовании не ведется, применение данного термина неправомочно.
Цитогенетическим методом исследуют различного рода гетероплоидию и хромосомные перестройки в соматических тканях человека, вызывающие различные фенотипические отклонения от нормы.
Чаще всего этот метод применяют на культуре ткани. Он позволяет учитывать крупные аномалии хромосом, возникающие как в половых, так и соматических клетках. Оказалось, что у человека, так же как и у животных, довольно часто возникают трисомики и моносомики по различным парам хромосом вследствие нерасхождения аутосом и половых хромосом в мейозе. Трисомия и моносомия по половым хромосомам у человека обнаруживаются на основе анализа полового хроматина.
В ходе относительно продолжительного индивидуального развития человека в клетках различных тканей накапливаются аномалии хромосом (хромосомные перестройки, а также изменение числа хромосом). Ткани организма представляют собой разнообразные популяции генетически различающихся клеток, в которых с возрастом концентрация клеток с патологическими ядрами возрастает. В этом случае цитогенетический метод позволяет изучать старение тканей на основе исследования структур клеток в возрастной динамике «популяции» соматических и генеративных тканей.
Поскольку частота возникновения хромосомных аномалий зависит от влияния на организм разнообразных мутагенов (ионизации, химических агентов - фармакологических препаратов, газового состава среды и др.), то цитогенетический метод позволяет устанавливать мутагенное действие факторов внешней среды на человека.
Применение цитогенетического метода особенно расширилось в связи с открытием причин ряда физических и психических заболеваний - так называемых хромосомных болезней.
Существует несколько заболеваний человека, например болезнь Клайнфельтера, Шерешевского-Тернера, Дауна и др., причины которых долго оставались неизвестными, пока цитологическими методами у таких больных не были обнаружены хромосомные аномалии.
Больные мужчины с синдромом Клайнфельтера характеризуются недоразвитием гонад, дегенерацией семенных канальцев, умственной отсталостью, непропорциональным ростом конечностей и т. д. У женщин встречается синдром Шерешевского-Тернера. Он проявляется в замедлении полового созревания, недоразвитии гонад, отсутствии менструаций, бесплодии, малом росте и в других Патологических признаках.
Оказалось, что оба эти синдрома у потомков являются следствием нерасхождения половых хромосом при образовании гамет родителей. Вследствие нерасхождения Х-хромосом у женского гомогаметного) пола в процессе мейоза могут возникать гаметы двумя Х-хромосомами, т. е. XX + 22 аутосомы, и без Х-хромосом, т. е. 0 + 22; у мужского (гетерогаметного) пола соответственно гаметы XY + 22 и 0 + 22. В случае оплодотворения таких яйцеклеток нормальными сперматозоидами (X + 22 или Y + 22) возможно образование следующих классов зигот: XXX + 44, 0Х + 44 и XXY + 44, 0Y + 44.
Из этого следует, что число хромосом у зигот разного происхождения может колебаться от 47 до 45, причем особи 0Y + 44, очевидно, не выживают, так как ни разу не были найдены. Хромосомный набор XXY + 44 присущ мужчине с синдромом Клайнфельтера (мужская интерсексуальность), хромосомные наборы Х0 + 44 и XXX + 44 имеют женщины с синдромом Шерешевского-Тернера.
При дальнейшем анализе больных с разными синдромами выяснилось, что вследствие нерасхождения половых хромосом могут возникать разного типа хромосомные аномалии, в частности полисомия. Встречаются, например, мужчины с такими наборами хромосом: XX Y, XXX Y, ХХХХ Y, а женщины - XXX, ХХХХ.
Особенность роли половых хромосом в детерминации пола у человека в случае их нерасхождения, в отличие от дрозофилы, проявилась в том, что набор хромосом XX Y всегда определяет мужской пол, а набор Х0 - женский. При этом увеличение числа Х-хромосом в сочетании с одной Y-хромосомой не изменяет определение мужского пола, а лишь усиливает синдром Клайнфельтера. Трисомия, или полисомия по Х-хромосоме, у женщин также часто вызывает заболевания, сходные с синдромом Шерешевского-Тернера.
Заболевания, вызванные нарушением нормального числа половых хромосом, диагностируются цитологическим методом - анализом полового хроматина. В тех случаях, когда в тканях мужчин имеется нормальный набор половых хромосом - XY, половой хроматин в клетках не обнаруживается. У нормальных женщин - XX - он обнаруживается в виде одного тельца. При полисомии по Х-хромосомам у женщин и мужчин количество телец полового хроматина всегда на единицу меньше числа Х-хромосом, т. е. n x = n·Х - 1. Так, в клетках мужчин с синдромом Клайнфельтера при наборе XX Y имеется одно тельце полового хроматина, при наборе XXXY - два, при наборе XXXXY - три; у женщин с синдромом Шерешевского-Тернера соответственно: Х0 - нет тельца, XXX - два тельца, ХХХХ - три тельца полового хроматина, и т. д. Предполагается, что в каждой такой зиготе генетически активна лишь одна из Х-хромосом. Остальные хромосомы переходят в гетеропикнотическое состояние в виде полового хроматина.
Причины этой закономерности не выяснены, однако предполагается, что она связана с нивелированием действия генов половых хромосом у гетеро- и гомогаметного пола.
Как мы знаем, нерасхождение хромосом может происходить не только в мейозе, но и в соматических клетках в течение всего эмбриогенеза животного начиная с первых дроблений яйца. В силу последнего среди людей при нарушении расхождения половых хромосом могут появиться больные мозаики-женщины и мозаики-мужчины. Так, например, описаны мозаики следующих типов: двойные: Х0/XX, Х0/XXX и X0/XY, X0/XYY, тройные: Х0/ХХ/ХХХ, XX/X0/XY, а также четверные мозаики, когда соматические клетки одного человека содержат четыре разных набора хромосом.
Кроме рассмотренного типа болезней, вызванных изменением числа половых хромосом в зиготе, хромосомные болезни могут быть вызваны нерасхождением аутосом и разного рода хромосомными перестройками (транслокациями, делециями). Так, например, у детей с врожденной идиотией - болезнью Дауна, сопровождающейся малым ростом, широким круглым лицом, близко расположенным узкими глазными щелями и полуоткрытым ртом, была обнаружена трисомия по 21 хромосоме. Установлено, что частота встречаемости болезни Дауна у новорожденных зависит от возраста матерей.
С врожденными хромосомными аномалиями связывают весьма разнообразные болезни. Поэтому цитогенетический метод приобретает важное значение в этиологии болезней человека.
Популяционный метод
Популяционный метод позволяет изучать распространение отдельных генов или хромосомных аномалий в человеческих популяциях.
Популяционный метод основывается на математических методах. Для анализа генетической структуры популяции необходимо обследовать большую по размеру выборку, которая должна быть репрезентативной - объективно отражать всю генеральную совокупность, т. е. всю популяцию в целом. В обследуемой выборке устанавливают распределение лиц по соответствующим четко очерченным фенотипическим классам, различия между которыми наследственно обусловлены. Затем, исходя из найденных фенотипических частот, определяют генные частоты.
На основе знания генных частот представляется возможность дать описание анализируемой популяции в соответствии с формулой Гарди-Вайнберга и заранее предсказать вероятный характер расщепления в потомстве лиц, относящихся к тем или иным фенотипическим классам. Исследование генных частот имеет важное значение для оценки последствий родственных браков, а также для выяснения генетической истории человеческой популяции в целом.
Частота распространения в популяциях разных аномалий оказывается различной; при этом подавляющее количество соответствующих рецессивных аллелей представлено в гетерозиготном состоянии.
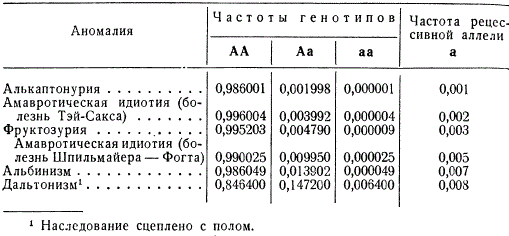
Так, примерно каждый сотый житель Европы гетерозиготен по гену амавротической идиотии (болезнь Шпильмайера-Фогта), тогда как заболевают этой болезнью в юношеском возрасте из 1 млн. только 25 человек, являющихся гомозиготными. Альбиносы в европейских странах встречаются с частотой 1 на 20 000, хотя гетерозиготное состояние этой аллели присуще каждому семидесятому жителю.
Несколько иначе дело обстоит в случае аномалий, наследующихся сцеплено с полом, примером чего может служить дальтонизм - цветная слепота, которая контролируется, по-видимому, рядом аллелей, распределенных по двум тесно сцепленным локусам в Х-хромосоме. Среди мужского населения частота дальтоников (q) соответствует суммарной частоте рецессивных аллелей и составляла, например, в Москве в 30-х годах, по данным Р. И. Серебровской, 7%, в то же время среди женского населения той же популяции цветная слепота была только у 0,5% (q 2), но в гетерозиготном состоянии примерно 13% женщин несут аллели, обусловливающие дальтонизм.
Как мы уже говорили выше, рассматривая генеалогический метод, вероятность появления в потомстве рецессивных гомозигот может быть различной при вступлении в брак лиц, имеющих разную степень родства. Так, у супругов, являющихся по отношению друг к другу двоюродными братьями и сестрами, вероятность рождения детей, гомозиготных по рецессивной аллели, распространенной в популяции с частотой q, составит уже не q 2 , а большую величину, а именно q/16 (1 +15q).
Это связано с тем, что если один из общих предков таких супругов - бабушка или дедушка - нес в гетерозиготе рецессивную аллель, то с вероятностью 1/16 данная аллель передастся обоим двоюродным сибсам.
Вредные последствия родственных браков особенно наглядно проявляются в изолированных популяциях ограниченного размера, так называемых изолятах . Под изолятом понимают группу особей популяции, которые вступают в брак большей частью с особями своей группы и поэтому характеризуются значительным коэффициентом кровного родства. Такими изолятами могут быть отдельные изолированные селения, общины и т. д. Внутри изолята более вероятны родственные браки (инбридинг), и больше шансов на то, что супруги будут нести одинаковые мутантные гены, следствием чего является увеличение вероятности проявления рецессивных аллелей в гомозиготном состоянии. Разные изоляты несут различные концентрации сходных или разных генов.
На Марианских островах и острове Гуам смертность среди местного населения от бокового амиотрофического склероза (связанного с поражением клеток передних рогов спинного мозга) в 100 с лишним раз превышает смертность от этой болезни в других странах. В Южной Панаме в провинции Сан-Блаз весьма заметную часть племени кариба куна составляют альбиносы, которые появляются здесь в каждом поколении. В одном селении на р. Роне в Швейцарии среди 2200 жителей имеется более 50 глухонемых, и еще у 200 обнаруживаются некоторые дефекты слуха. По всей вероятности, во всех подобных случаях резкого увеличения концентрации отдельных аллелей известную роль играет генетический дрифт, неравномерное размножение в прошлом отдельных семей, родов, а также снижение миграции.
По мере роста цивилизации и развития производительных сил общества количество изолятов уменьшается, и их значение для популяции в целом падает. Однако они все еще имеют место.
Знание генных частот, как уже говорилось позволяет предсказывать характер расщепления в потомстве отдельных фенотипических классов родительских особей.
Исходя из формулы Гарди-Вайнберга, можно показать, что при моногенном наследовании расщепление по фенотипу в потомстве доминантных матерей должно осуществляться в соотношении p(1 + pq) доминантов к р рецессивов, или (l+pq):q 2 ; в потомстве рецессивных матерей расщепление по фенотипу должно быть pq 2: q 3 , или p: q.
Приведем пример. В одном исследовании при изучении резус-фактора частота рецессивной аллели rh в популяции составила 0,4, а частота доминантной аллели Rh - 0,6. Отсюда следовало ожидать, что в потомстве резус-положительных матерей частота резус — положительных детей (Rh +) примерно в 7,8 раза будет превышать частоту резус-отрицательных детей (Rh —); в потомстве резус-отрицательных матерей соответствующее превышение будет в 1,5 раза.
Действительные соотношения в обследованной выборке составили:
- в первом случае 1475 Rh + : 182 Rh — , или 8,1: 1,
- во втором случае 204 Rh + : 129 Rh — , или 1,6: 1.
Таким образом, наблюдаемые результаты при расщеплении весьма хорошо соответствуют теоретически ожидаемым результатам, предсказанным на основе анализа генных частот.
Популяционный анализ полиморфизма по группам крови интересен тем, что он помогает понять динамику генетической структуры различных популяций и способствует выявлению связей между ними.
Разные популяции существенно различаются по своей генетической структуре, в частности по группам крови. При этом удается проследить некоторые вполне четкие закономерности. Если концентрация аллели I B наибольшая в районе Индии и Китая, то к востоку и западу от этого района происходит постепенное падение ее вплоть до нуля среди коренных обитателей Америки и Австралии. В то же время у американских индейцев (и аборигенов Австралии и Полинезии) максимума достигает концентрация аллели I 0 . Аллель I А редка у коренного населения Америки, а также в Индии, Аравии, тропической Африке, в Западной Европе.
Для объяснения этих различий в генетической структуре популяций недавно была предложена гипотеза, согласно которой решающим фактором отбора в отношении групп крови системы АВ0 явились эпидемии чумы и оспы. Возбудитель чумы Pasteuvella pest is, обладая свойством антигена 0, оказывается наиболее губительным для людей с группой крови 0, поскольку такие лица не способны вырабатывать достаточное количество антител в случае инфекции. По аналогичной причине вирус оспы наиболее опасен для людей с группой крови А. Там, где свирепствовала чума (Индия, Монголия, Китай, Египет), шла интенсивная элиминация аллели I 0 , а там, где особенно свирепствовала оспа (Америка, Индия, Аравия, тропическая Африка), в первую очередь элиминировалась аллель 1 А. В районах Азии, где чума и оспа были эндемичны, наибольшую частоту получила аллель 1 в.
В главе 5 мы рассмотрели моногенное наследование серповидноклеточной анемии, обусловленное расщеплением по аллелям гена S. Высокая концентрация аллели S в поясе эндемичной малярии (Африка, Средиземноморье) оказалась связанной с повышенной устойчивостью к малярии гетерозигот (Ss) и с возникновением. в результате этого системы сбалансированного наследственного полиморфизма.
Таким образом, в обоих приведенных примерах анализа полиморфизма по группам крови и серповидно-клеточной анемии мы видим, как применение популяционного метода позволяет вскрывать генетическую структуру человеческих популяций.
Онтогенетический метод
Онтогенетический метод позволяет устанавливать по фенотипу носительство рецессивных аллелей в гетерозиготном состоянии и хромосомных перестроек.
Генетической основой проявления рецессивных генов в гетерозиготном состоянии является, по-видимому, неполный блок в цепи синтеза того или иного метаболита, вызванного действием доминантной аллели данного гена.
Известно, что некоторые наследственные болезни проявляются не только у лиц, гомозиготных по аллелям, вызывающим заболевание, но в стертой форме и у гетерозигот. Поэтому в настоящее время усиленно разрабатываются методы определения гетерозиготного носительства в онтогенезе. Так, гетерозиготный носитель фенилкетонурии (повышенное содержание фенилаланина в крови определяется дополнительным введением фенилаланина и последующим определением уровня его (или тирозина) в плазме крови. Наличие гетерозиготности по данной аллели устанавливается по повышенному содержанию фенилаланина. В норме (т. е. у гомозигот по доминантной аллели) уровень фенилаланина не изменяется. В норме в крови присутствует фермент каталаза, необходимый для углеводного обмена, но встречается ген, который в гомозиготном состоянии вызывает отсутствие каталазы. У гомозиготных носителей этого гена наблюдается болезнь акаталаземия - расстройство углеводного обмена. Гетерозиготы занимают промежуточное положение по активности каталазы без большого захождения между доминантными и рецессивными гомозиготами.
По активности каталазы можно точно определить гетерозиготных и гомозиготных носителей аллели акаталаземии среди близких родственников и родителей.
Гетерозиготное носительство аллели, определяющей мышечную дистрофию типа Дюшена, тестируется по активности криатинфосфокиназы. Теперь разработаны, подобные тесты для 40 наследственных болезней, определяемых рецессивными аллелями.
В настоящее время онтогенетический метод обогатился за счет биохимических, иммунологических и молекулярных приемов исследования, описанию которых посвящен ряд специальных руководств.
Важность онтогенетического метода очевидна для установления носительства рецессивного гена в гетерозиготном состоянии у родственников семьи, в которой появляется наследственно больной ребенок. Диагностика в онтогенезе важна для расчета вероятности появления наследственно больных потомков при родственных и смешанных браках. По мере упрощения тестирования гетерозиготного носительства этот метод должен будет внедряться в целях консультации супружеских пар относительно возможности появления заболевания у их детей, а также для изучения распространения мутаций в популяциях.
Если вы нашли ошибку, пожалуйста, выделите фрагмент текста и нажмите Ctrl+Enter .
Для генетических исследований человек является неудобным объектом, так как у человека: невозможно экспериментальное скрещивание; большое количество хромосом; поздно наступает половая зрелость; малое число потомков в каждой семье; невозможно уравнивание условий жизни для потомства.
В генетике человека используется ряд методов исследования.
Генеалогический метод
Использование этого метода возможно в том случае, когда известны прямые родственники — предки обладателя наследственного признака (пробанда ) по материнской и отцовской линиям в ряду поколений или потомки пробанда также в нескольких поколениях. При составлении родословных в генетике используется определенная система обозначений. После составления родословной проводится ее анализ с целью установления характера наследования изучаемого признака.
Условные обозначения, принятые при составлении родословных:
1 — мужчина; 2 — женщина; 3 — пол не выяснен; 4 — обладатель изучаемого признака; 5 — гетерозиготный носитель изучаемого рецессивного гена; 6 — брак; 7 — брак мужчины с двумя женщинами; 8 — родственный брак; 9 — родители, дети и порядок их рождения; 10 — дизиготные близнецы; 11 — монозиготные близнецы.
Благодаря генеалогическому методу были определены типы наследования многих признаков у человека. Так, по аутосомно-доминантному типу наследуются полидактилия (увеличенное количество пальцев), возможность свертывать язык в трубочку, брахидактилия (короткопалость, обусловленная отсутствием двух фаланг на пальцах), веснушки, раннее облысение, сросшиеся пальцы, заячья губа, волчья пасть, катаракта глаз, хрупкость костей и многие другие. Альбинизм, рыжие волосы, подверженность полиомиелиту, сахарный диабет, врожденная глухота и другие признаки наследуются как аутосомно-рецессивные.

Доминантный признак — способность свертывать язык в трубочку (1) и его рецессивный аллель — отсутствие этой способности (2).
3 — родословная по полидактилии (аутосомно-доминантное наследование).
Целый ряд признаков наследуется сцепленно с полом: Х -сцепленное наследование — гемофилия, дальтонизм; Y -сцепленное — гипертрихоз края ушной раковины, перепончатость пальцев ног. Имеется ряд генов, локализованных в гомологичных участках Х - и Y -хромосом, например общая цветовая слепота.
Использование генеалогического метода показало, что при родственном браке, по сравнению с неродственным, значительно возрастает вероятность появления уродств, мертворождений, ранней смертности в потомстве. В родственных браках рецессивные гены чаще переходят в гомозиготное состояние, в результате развиваются те или иные аномалии. Примером этого является наследование гемофилии в царских домах Европы.

— гемофилик; — женщина-носитель.
Близнецовый метод

1 — монозиготные близ-нецы; 2 — дизигот-ные близ-нецы.
Близнецами называют одновременно родившихся детей. Они бывают монозиготными (однояйцевыми) и дизиготными (разнояйцевыми).
Монозиготные близнецы развиваются из одной зиготы (1), которая на стадии дробления разделилась на две (или более) части. Поэтому такие близнецы генетически идентичны и всегда одного пола. Монозиготные близнецы характеризуются большой степенью сходства (конкордантностью ) по многим признакам.
Дизиготные близнецы развиваются из двух или более одновременно овулировавших и оплодотворенных разными сперматозоидами яйцеклеток (2). Поэтому они имеют различные генотипы и могут быть как одного, так и разного пола. В отличие от монозиготных, дизиготные близнецы характеризуются дискордантностью — несходством по многим признакам. Данные о конкордантности близнецов по некоторым признакам приведены в таблице.
| Признаки | Конкордантность, % | |
|---|---|---|
| Монозиготные близнецы | Дизиготные близнецы | |
| Нормальные | ||
| Группа крови (АВ0) | 100 | 46 |
| Цвет глаз | 99,5 | 28 |
| Цвет волос | 97 | 23 |
| Патологические | ||
| Косолапость | 32 | 3 |
| «Заячья губа» | 33 | 5 |
| Бронхиальная астма | 19 | 4,8 |
| Корь | 98 | 94 |
| Туберкулез | 37 | 15 |
| Эпилепсия | 67 | 3 |
| Шизофрения | 70 | 13 |
Как видно из таблицы, степень конкордантности монозиготных близнецов по всем приведенным признакам значительно выше, чем у дизиготных, однако она не является абсолютной. Как правило, дискордантность монозиготных близнецов возникает в результате нарушений внутриутробного развития одного из них или под влиянием внешней среды, если она была разной.
Благодаря близнецовому методу, была выяснена наследственная предрасположенность человека к ряду заболеваний: шизофрении, эпилепсии, сахарному диабету и другим.
Наблюдения за монозиготными близнецами дают материал для выяснения роли наследственности и среды в развитии признаков. Причем под внешней средой понимают не только физические факторы среды, но и социальные условия.
Цитогенетический метод
Основан на изучении хромосом человека в норме и при патологии. В норме кариотип человека включает 46 хромосом — 22 пары аутосом и две половые хромосомы. Использование данного метода позволило выявить группу болезней, связанных либо с изменением числа хромосом, либо с изменениями их структуры. Такие болезни получили название хромосомных .
Материалом для кариотипического анализа чаще всего являются лимфоциты крови. Кровь берется у взрослых из вены, у новорожденных — из пальца, мочки уха или пятки. Лимфоциты культивируются в особой питательной среде, в состав которой, в частности, добавлены вещества, «заставляющие» лимфоциты интенсивно делиться митозом. Через некоторое время в культуру клеток добавляют колхицин. Колхицин останавливает митоз на уровне метафазы. Именно во время метафазы хромосомы являются наиболее конденсированными. Далее клетки переносятся на предметные стекла, сушатся и окрашиваются различными красителями. Окраска может быть а) рутинной (хромосомы окрашиваются равномерно), б) дифференциальной (хромосомы приобретают поперечную исчерченность, причем каждая хромосома имеет индивидуальный рисунок). Рутинная окраска позволяет выявить геномные мутации, определить групповую принадлежность хромосомы, узнать, в какой группе изменилось число хромосом. Дифференциальная окраска позволяет выявить хромосомные мутации, определить хромосому до номера, выяснить вид хромосомной мутации.
В тех случаях, когда необходимо провести кариотипический анализ плода, для культивирования берутся клетки амниотической (околоплодной) жидкости — смесь фибробластоподобных и эпителиальных клеток.
К числу хромосомных заболеваний относятся: синдром Клайнфельтера, синдром Тернера-Шерешевского, синдром Дауна, синдром Патау, синдром Эдвардса и другие.
Больные с синдромом Клайнфельтера (47, ХХY ) всегда мужчины. Они характеризуются недоразвитием половых желез, дегенерацией семенных канальцев, часто умственной отсталостью, высоким ростом (за счет непропорционально длинных ног).
Синдром Тернера-Шерешевского (45, Х0 ) наблюдается у женщин. Он проявляется в замедлении полового созревания, недоразвитии половых желез, аменорее (отсутствии менструаций), бесплодии. Женщины с синдромом Тернера-Шерешевского имеют малый рост, тело диспропорционально — более развита верхняя часть тела, плечи широкие, таз узкий — нижние конечности укорочены, шея короткая со складками, «монголоидный» разрез глаз и ряд других признаков.
Синдром Дауна — одна из самых часто встречающихся хромосомных болезней. Она развивается в результате трисомии по 21 хромосоме (47; 21, 21, 21). Болезнь легко диагностируется, так как имеет ряд характерных признаков: укороченные конечности, маленький череп, плоское, широкое переносье, узкие глазные щели с косым разрезом, наличие складки верхнего века, психическая отсталость. Часто наблюдаются и нарушения строения внутренних органов.
Хромосомные болезни возникают и в результате изменения самих хромосом. Так, делеция р -плеча аутосомы №5 приводит к развитию синдрома «крик кошки». У детей с этим синдромом нарушается строение гортани, и они в раннем детстве имеют своеобразный «мяукающий» тембр голоса. Кроме того, наблюдается отсталость психомоторного развития и слабоумие.
Чаще всего хромосомные болезни являются результатом мутаций, произошедших в половых клетках одного из родителей.
Биохимический метод
Позволяет обнаружить нарушения в обмене веществ, вызванные изменением генов и, как следствие, изменением активности различных ферментов. Наследственные болезни обмена веществ подразделяются на болезни углеводного обмена (сахарный диабет), обмена аминокислот, липидов, минералов и др.
Фенилкетонурия относится к болезням аминокислотного обмена. Блокируется превращение незаменимой аминокислоты фенилаланин в тирозин, при этом фенилаланин превращается в фенилпировиноградную кислоту, которая выводится с мочой. Заболевание приводит к быстрому развитию слабоумия у детей. Ранняя диагностика и диета позволяют приостановить развитие заболевания.
Популяционно-статистический метод
Это метод изучения распространения наследственных признаков (наследственных заболеваний) в популяциях. Существенным моментом при использовании этого метода является статистическая обработка получаемых данных. Под популяцией понимают совокупность особей одного вида, длительное время обитающих на определенной территории, свободно скрещивающихся друг с другом, имеющих общее происхождение, определенную генетическую структуру и в той или иной степени изолированных от других таких совокупностей особей данного вида. Популяция является не только формой существования вида, но и единицей эволюции, поскольку в основе микроэволюционных процессов, завершающихся образованием вида, лежат генетические преобразования в популяциях.
Изучением генетической структуры популяций занимается особый раздел генетики — популяционная генетика . У человека выделяют три типа популяций: 1) панмиктические, 2) демы, 3) изоляты, которые отличаются друг от друга численностью, частотой внутригрупповых браков, долей иммигрантов, приростом населения. Население крупного города соответствует панмиктической популяции. В генетическую характеристику любой популяции входят следующие показатели: 1) генофонд (совокупность генотипов всех особей популяции), 2) частоты генов, 3) частоты генотипов, 4) частоты фенотипов, система браков, 5) факторы, изменяющие частоты генов.
Для выяснения частот встречаемости тех или иных генов и генотипов используется закон Харди-Вайнберга .
Закон Харди-Вайнберга
В идеальной популяции из поколения в поколение сохраняется строго определенное соотношение частот доминантных и рецессивных генов (1), а также соотношение частот генотипических классов особей (2).
p + q = 1, (1)р 2 + 2pq + q 2 = 1, (2)
где p — частота встречаемости доминантного гена А ; q — частота встречаемости рецессивного гена а ; р 2 — частота встречаемости гомозигот по доминанте АА ; 2pq — частота встречаемости гетерозигот Аа ; q 2 — частота встречаемости гомозигот по рецессиву аа .
Идеальной популяцией является достаточно большая, панмиктическая (панмиксия — свободное скрещивание) популяция, в которой отсутствуют мутационный процесс, естественный отбор и другие факторы, нарушающие равновесие генов. Понятно, что идеальных популяций в природе не существует, в реальных популяциях закон Харди-Вайнберга используется с поправками.
Закон Харди-Вайнберга, в частности, используется для примерного подсчета носителей рецессивных генов наследственных заболеваний. Например, известно, что в данной популяции фенилкетонурия встречается с частотой 1:10000. Фенилкетонурия наследуется по аутосомно-рецессивному типу, следовательно, больные фенилкетонурией имеют генотип аа , то есть q 2 = 0,0001. Отсюда: q = 0,01; p = 1 - 0,01 = 0,99. Носители рецессивного гена имеют генотип Аа , то есть являются гетерозиготами. Частота встречаемости гетерозигот (2pq ) составляет 2 · 0,99 · 0,01 ≈ 0,02. Вывод: в данной популяции около 2% населения — носители гена фенилкетонурии. Заодно можно подсчитать частоту встречаемости гомозигот по доминанте (АА ): p 2 = 0,992, чуть меньше 98%.
Изменение равновесия генотипов и аллелей в панмиктической популяции происходит под влиянием постоянно действующих факторов, к которым относятся: мутационный процесс, популяционные волны, изоляция, естественный отбор, дрейф генов, эмиграция, иммиграция, инбридинг. Именно благодаря этим явлениям возникает элементарное эволюционное явление — изменение генетического состава популяции, являющееся начальным этапом процесса видообразования.
Генетика человека — одна из наиболее интенсивно развивающихся отраслей науки. Она является теоретической основой медицины, раскрывает биологические основы наследственных заболеваний. Знание генетической природы заболеваний позволяет вовремя поставить точный диагноз и осуществить нужное лечение.
Перейти к лекции №21 «Изменчивость»